
{kind=link}
【研究背景】
传统工业上的合成氨技术涉及高能耗、污染大的Haber-Bosch工艺,电催化氮还原(NRR)技术为绿色合成氨提供了新途径。然而,在水溶液中,由于存在竞争反应——析氢反应(HER),以及电催化剂对氮的利用率较低,使得NRR的活性与选择性较低。
【成果介绍】
yl7703永利官网晏成林教授课题组报道,通过利用高浓度电解质中的盐析效应,可以同时解决NRR的活性与选择性低的难题,并实现氨的高效合成。研究发现,在高浓度电解液中,溶质离子对周围的水分子表现出很强的亲和力,形成水合壳层,从而限制了它们作为质子源和溶剂的作用。这不仅有效地抑制了HER,而且通过沉淀物的异相成核来实现反应界面上N2的富集,因而可以提高NRR的活性和选择性。即使使用无金属电催化剂来催化NRR,其在-0.3 V下法拉第效率也可达到71±1.9%。该工作以Salting-out effect promoting highly efficient ambient ammonia synthesis为题在Nature Communications上发表论文。
无独有偶,晏成林教授课题组在上个月(4月19日)于Nature Catalysis上发表题为Proton-filtering covalent organic frameworks with superior nitrogen penetration flux promote ambient ammonia synthesis 的论文。该工作通过使用H+过滤的ECOF层来抑制竞争性HER反应物的扩散过程,同时利用该膜对氮分子良好的亲和力和N2渗透通量性质,最终使得ECOF层修饰表面的BCP催化剂表现出优异的NRR活性与选择性。详情请见推文:https://mp.weixin.qq.com/s/Z3QcPiZl8fj-nFvdyDw8Vg
【图文介绍】
{kind=link}
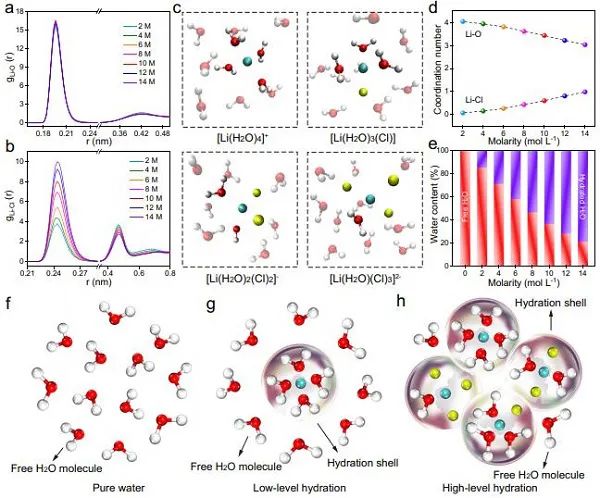
图1 水合作用体系的分子动力学模拟:(a)Li+-O、(b)Li+-Cl−相互作用的径向分布函数;(c) Li水合结构在四种稳定团簇中保持平衡;(d)Li+-O和Li+-Cl–的配位数随LiCl浓度的变化;(e)不同浓度LiCl溶液中的含水量;(f-h)水合壳在纯水、低水合体系以及高水合体系中的示意图。
LiCl在水中溶解性好,且可以产生较强的水合作用,因此选择LiCl作为本研究采用的锂盐。通过分子动力学(MD)模拟,研究了不同LiCl浓度(2-14 M)下水合作用体系的结构和动力学性质。首先考虑了Li+-H2O相互作用,其径向分布函数(RDFs)如图1a所示。Li+由于其体积小、电荷密度高,通常与周围的H2O分子发生强烈相互作用,从而形成第一级水合球,这与RDFs位于0.196 nm处出现尖锐Li+-O峰相对应。在大约0.42 nm处的峰比较平坦,因此在本工作中并不考虑Li+的第二级水合球。
接着考虑了Li+-Cl–相互作用,图1b的RDFs在0.244 nm处所出现的峰证实了Li+-Cl–相互作用。随着盐浓度的增加,这个信号会进一步增强,即更多的Cl–会穿透Li的水合球,取代一些水分子。Li水合球在[Li+(H2O)4]+、[Li+(H2O)3Cl–]、[Li+(H2O)2(Cl)2]–和[Li+(H2O)(Cl)3]2–四个稳定团簇之间保持平衡。如图1c的快照所示,Li离子的总配位数均为4。
随着锂盐浓度的增加,Li+的水合配位数略有下降(图1d),但水合作用的水分子总数继续增加,即游离水不断减少(图1e)。也就是说,与纯水相比,盐的浓度会影响质子的活度(图1f)。在低水合体系中,少量的Li水合壳均匀分布在体系中,此时自由H2O分子数量仍较高(图1g),因此该体系仍可保证质子的充足供给,仍然有利于HER的进行。当LiCl浓度进一步增加时,参与Li水合球的H2O分子数量增加,游离H2O较少(图1h),质子供给受抑制。
{kind=link}
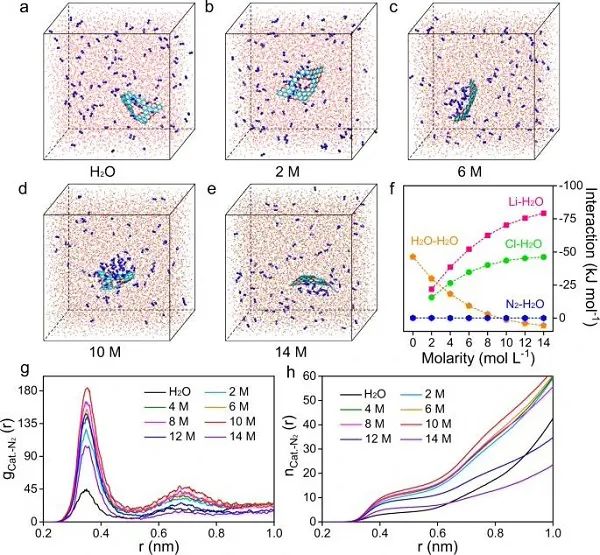
图2 多相催化体系的分子动力学模拟:(a-e)不同LiCl浓度的多相催化体系在20 ns下的快照;(f)多相催化体系中离子-水、水-水、气-水的相互作用能;催化剂表面N2的(g)径向分布函数以及(h)其积分函数。
基于上述结果,可以认为LiCl是诱发盐析效应的理想溶质。因此,作者随后研究了LiCl对多相催化体系的影响。不同锂盐浓度的快照如图2a-e所示。显然,与纯水相比,高浓度溶液中电解质/电催化剂界面处存在N2富集现象。通过计算了体系内的一些相互作用,以探索潜在的机制,如图2f所示,在纯水中,只存在H2O-H2O相互作用和弱的N2-H2O相互作用。N2分子保持动态平衡状态,在氢键网络中均匀分布,没有形成局部浓度差。在LiCl溶液中,Li+、Cl–和N2分子均与H2O分子相互作用。由于N2具有微小的四极矩,它对H2O的亲和力总是最低的。另一方面,在2-14 M LiCl溶液中,Li+-H2O相互作用和Cl–-H2O相互作用都更加强烈。
为了定量研究N2的富集效应,作者得到了如图2g-h的表面N2的RDFs以及其积分函数。显然,在相同距离内,在高浓度电解液中,与纯水相比,电催化剂周围的N2分子密度要高得多,这种N2富集效应在10 M LiCl中最明显。
{kind=link}
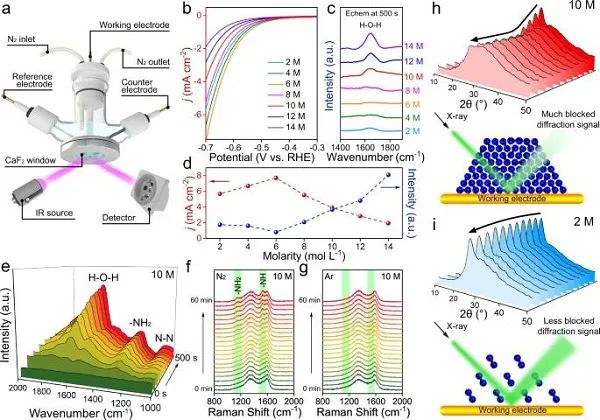
图3 HER性能和原位表征:(a)用于原位FTIR表征的特制电解池;(b)饱和Ar环境下不同体系的LSV曲线;(c)在-0.3 V下的原位FTIR谱图;(d)HER活性与FTIR谱图中H-O-H弯曲振动强度随锂盐浓度的变化;(e)在-0.3 V下、电解质为10 M LiCl的原位FTIR谱图随时间变化;(f,g)在-0.3 V下、含饱和N2或饱和Ar的10 M LiCl的原位拉曼光谱随时间变化;(h,i) 在-0.3 V下、以10 M或2 M LiCl为电解液的原位XRD图与相应的机理。
为了探究该催化剂对HER的抑制是否在实际催化过程中起作用,在一个特制的装置中对不同体系进行了原位FTIR表征(图3a)。首先在饱和Ar环境下测量了LSV曲线,如图3b所示。有趣的是,HER活性在LiCl浓度为2-6 M下略有增加,然后在LiCl浓度为8-14 M下发生衰减。对应的原位FTIR光谱显示H2O分子的H-O-H弯曲振动强度与HER活性呈相反的趋势,如图3c、d所示,这一现象与实验结果相吻合。在2-6 M LiCl中,工作电极周围的H2O分子被电解,红外光谱中可捕获的自由H2O分子较少。随着锂盐浓度进一步增加,HER逐渐被抑制,更多的H2O分子停留在电极表面,H-O-H弯曲振动强度增大。通过结合电化学测试和原位表征,证实了高浓度的LiCl溶液可用于有效抑制HER。
作者进一步通过多种原位技术来观察NRR。MD和有限元模拟表明,LiCl为10 M时,Stern层的N2积累量最大。如图3e所示,FTIR谱图在1669 cm−1处吸收峰对应H2O的H-O-H弯曲振动,在1298 cm−1和1090 cm−1处的两个吸收峰分别可归为吸附的N2Hy的-NH2弯曲和N-N拉伸振动。在饱和N2氛围下,原位拉曼光谱显示,位于1152和1526 cm–1的两个峰,分别对应-NH2和-NH(图3f),而在饱和Ar氛围中没有观察到这种现象(图3g)。
为了进一步验证盐析效应对工作电极上N2吸附的影响,对不同浓度LiCl溶液体系进行了原位XRD表征。对于10 M LiCl(图3h),工作电极在初始条件下所观察到对应碳的主要衍射峰(约26.5°处),并在初始工作前半小时内逐渐减弱。这可能是由于N2的连续吸附,从而在XRD表征过程中干扰了部分碳信号。随着反应的进行,N2的吸附和消耗达到平衡,碳信号在接下来的半小时内保持稳定。相比之下,当使用2 M LiCl作为电解液时(图3i),由于它在电极表面诱导N2富集的能力较弱,在XRD谱图中观察到的碳衍射峰变化较小。因此,电解液中锂盐浓度要足够高,才能通过盐析效应来实现N2在电极表面的积累。而稀溶液仅在电极表面累积少量的N2,不足以实现高效的NRR过程。
{kind=link}
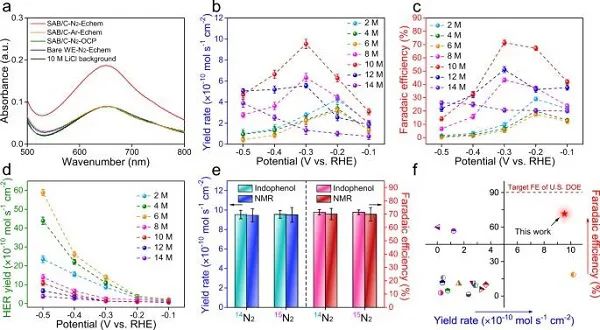
图4 室温下电催化氮还原合成氨:(a)电解质在不同条件下的紫外-可见吸收光谱;(b-d)不同体系的NH3产率、对应的法拉第效率、析氢速率;(e)在−0.3 V下、采用不同进料气体时,采用吲哚酚法和NMR法分别测得法拉第效率和NH3产率;(f)与其他非贵金属电催化剂的NH3产率和法拉第效率对比。
使用气密H型电解槽来测试NRR活性,通过比较电解质在不同条件下含NH3量,并用紫外-可见吸收光谱进行检测,如图4a所示,揭示了NH3的合成来源于N2的电化学还原。
通过计时安培测试对不同体系的NRR性能进行定量分析。图4b-c显示了在-0.1 ~ -0.5 V 下NH3的产率以及对应的法拉第效率。结果表明,在2 ~ 6 M LiCl中,NH3产率和法拉第效率较低。随着锂盐浓度的进一步增加,HER活性逐渐被抑制,质子更倾向于被吸附的N2进行加氢。使用10 M LiCl为电解质时,即使使用无金属电催化剂,在-0.3 V下法拉第效率也高达71±1.9%,NH3产率为(9.5±0.4)×10–10 mol s–1 cm–2。
此外,通过吲哚酚法和NMR法,对NH3产率与对应的法拉第效率进行比较,如图4e所示,说明了实验结果的可靠性。图4f显示,即使与最先进的无贵金属电催化剂相比,本工作在NH3产率、法拉第效率方面表现突出,非常接近美国能源部在ARPA-E REFUEL计划所设定的目标。
【总结与展望】
本工作强调了盐析效应对提高NRR效率的重要性,并合理利用该效应,在室温下同时提高了NRR的选择性和活性。在稀溶液中,水合壳的数量有限,而自由水的数量仍然很高。与稀溶液相比,高浓度的盐溶液会破坏或扭曲氢键网络,极大地增加了溶质离子周围固定水分子的密度。因此,N2分子周围可用自由水的密度显著降低,迫使它们从溶液中沉淀并逐渐在电解质/电催化剂界面上积累。因此,以高浓度LiCl溶液为电解质时,可以在Stern层内实现理想的N2富集以及适量的质子供给,这进一步由分子动力学模拟、有限元模拟以及各种原位表征加以佐证。
因此,使用10 M LiCl为电解质时,即使使用无金属电催化剂,在-0.3 V下法拉第效率也高达71±1.9%,NH3产率为(9.5±0.4)×10–10mol s–1cm–2,接近美国能源部ARPA-E REFUEL计划所设定的目标。本文所提出的策略可以应用于其他电化学系统,能有效抑制竞争反应,同时实现进料气体在反应界面上富集。
【文献信息】
题目:Salting-out effect promoting highly efficient ambient ammonia synthesis
DOI:10.1038/s41467-021-23360-0
链接:https://www.nature.com/articles/s41467-021-23360-0